Binghui Shen, Ph.D.


Our laboratory focuses on understanding the cellular mechanisms that maintain and disrupt genomic stability, contributing to many diseases, particularly cancer. The genome is especially vulnerable during DNA replication. Our lab studies the mechanisms underlying DNA replication and DNA damage response and repair in normal and cancer cells using multidisciplinary techniques, including biochemical, cellular, structural biological, and chemical approaches and genetic mouse modeling. Our latest research includes:
Structure-specific nuclease FEN1
For many years, one of the most pressing questions regarding structure-specific nucleases was which structural elements enable these proteins to recognize DNA/RNA substrates based on their structural conformation rather than on nucleic acid sequence motifs. In collaboration with Dr. John Tainer, we determined the three-dimensional crystallographic structures of the FEN1 protein, the FEN1/PCNA complex, and the FEN1/DNA substrate complex. We made more than 300 rational mutations to identify the key structural elements that facilitate substrate recognition, binding, cleavage, and dissociation.
Our lab also established clinically relevant mouse models that carry FEN1 mutations found in human cancer patients to identify the etiological roots of cancer. We inbred fourteen mouse lines harboring various categories of FEN1 mutations, including those that affect segregation of nuclease activities, impair protein/protein interactions with PCNA or WRN, and eliminate post-translational modification sites. These mouse lines were predisposed to cancers, and their use has provided a tremendous amount of information regarding the mechanisms by which FEN1 mutations lead to cancers.
Another key question in the DNA replication/repair field is how sequential enzymatic reactions, including Pol δ-driven gap-filling, FEN1-mediated RNA flap cleavage, and Lig I-mediated DNA ligation, occur in the proper order at DNA replication forks. Each enzyme must be recruited to the replication site at the correct time and, after the reaction is complete, must promptly dissociate from the DNA substrate. PCNA has been implicated as the platform for coordinating these sequential actions, and our lab has worked on elucidating how PCNA/FEN1 interactions and FEN1 modifications contribute to this process. Using mass spectrometry, we identified the interaction partners of FEN1, including protein modifying enzymes such as protein methyltransferases, kinases, and ubiquitin ligases. We also discovered that, whereas methylation of FEN1 places it onto the DNA replication fork, phosphorylation dissociates it during the progression of the S-phase, and sequential post-translational modifications program the degradation of FEN1 at the end of the S-phase. Furthermore, we found that disordering these sequential reactions impairs cell cycle programs. This work has been the subject of many commentaries and has opened new research directions.
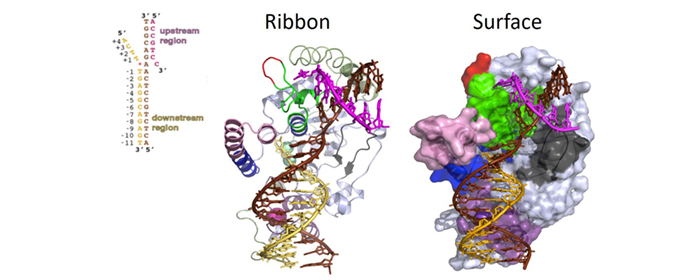
Flap endonuclease 1 (FEN1)
The DNA2 in counteracting DNA replication stresses
Our group was the first to report that the nuclear DNA-encoded nuclease DNA2 predominantly migrates to mitochondria and plays an important role in mitochondrial DNA replication and oxidative DNA damage repair. Although this surprising result was somewhat controversial, it drove other groups to look for DNA2 mutations in families with mitochondrial disease, which led to the finding that mutations in DNA2 cause mitochondrial-based myopathy. In later studies, we showed that DNA2 only localizes to the nucleus under DNA replication stress, such as when processing difficult-to-separate sequences. Using established mouse models and cell lines, we found that DNA2 is required to maintain the integrity of telomeres and resolve G-quadruplexes.
We recently demonstrated that DNA2 predominantly binds to centromeric DNA regions and that loss of DNA2 results in stalled replication of centromeres. These centromeric DNA replication defects activate the DNA damage checkpoint kinase ataxia telangiectasia and Rad3-related protein (ATR), contributing to cell cycle arrest in the late‐S/G2 phase. Furthermore, DNA2 nuclease‐ or helicase‐deficient cells exhibit compromised loading of centromere protein A (CENP‐A) onto chromatin and loss of intact centromeric DNA. In addition, cells that escape the G2/M checkpoint showed the inappropriate formation of the metaphase plate and chromosomal missegregation.
Considering that DNA2 is highly overexpressed in breast and other cancer cells, our group has developed inhibitors to advance the study of DNA2 and potential new therapeutic agents.
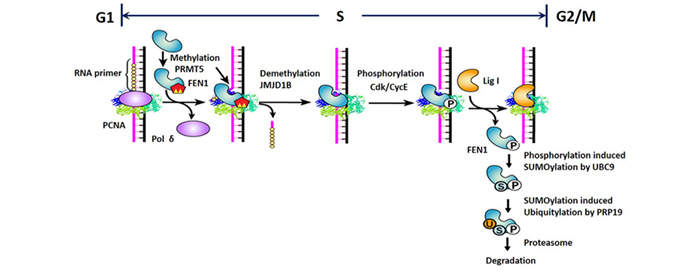
Posttranslational modifications determine the actions of FEN1 in DNA replication fork
Nature Chem. Biol. 2010 | Mol. Cell 2012
Genetic signature of prostate cancer
Prostate cancer (PCa) is one of the most heritable human cancers, suggesting a significant contribution from genetic risk factors. However, the underlying genetic architecture of PCa remains largely unknown. Despite implicating ~100 statistically validated loci in PCa risk, recent genome-wide association studies have provided little explanation for the expected heritability. We hypothesize that the disease susceptibility loci possess rare and functionally deficient variants responsible for PCa risk, similar to the “loss-of-function” mutations identified in the BRCA1/2 loci in breast and ovarian cancers. We will take advantage of a large collection of family PCa specimens to identify the genetic factors, including missense exon and non-coding RNA mutations that account for PCa. We will then use our expertise in functional analysis to determine how these mutations contribute to cancer formation (e.g., through PCa-specific genome instability).
We expect that elucidating the genetic architecture of PCa, including information regarding the mechanisms by which PCa-specific genome instability originates and contributes to cancer formation, will guide us to design better preventative and therapeutic treatments for PCa.
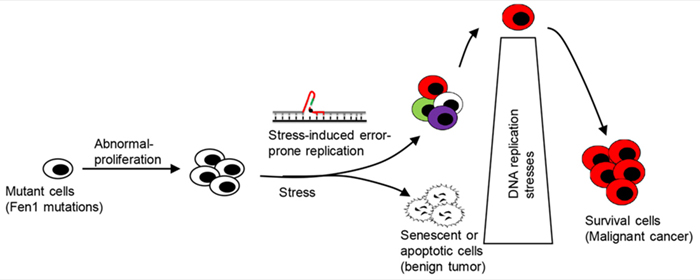
How cancer initiation cells escape barriers of DNA replication stress?
Nature Med. 2007 | Nature Comms. 2012 | Science 2021
Duarte Cancer Center
Duarte, CA 91010