Vaidehi Lab Research
Laboratory Overview
Proteins are allosteric nanomachines whose conformational dynamics confer their functional versatility. Therefore, it is important to map the conformational dynamics of proteins to:
- Understand the mechanism of allosteric communication.
- Understand how the same protein modulates its shape to perform different functions.
- Design drugs with functional specificity.
- Identify allosteric-druggable sites on the surface of proteins.
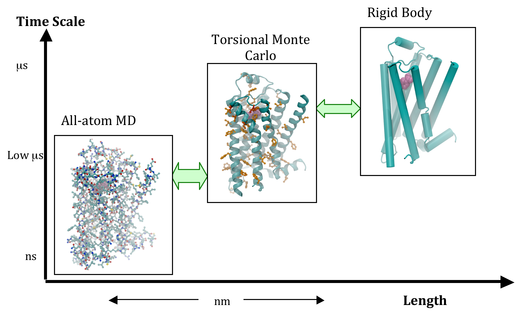
Our lab is focused on developing physics-based, state-of-the-art multiscale computational methods to study the conformational dynamics of proteins and in drug design. One of the major bottlenecks in using the existing computational methods to study dynamics of proteins is the limitation in time scale and the narrow conformational search afforded by these methods. Thus, we need multiscale computational methods that span a longer range in time and length scale to be able to simulate biological processes. We are developing coarse-grain computational methods to sample the various kinetic states of the protein dynamics, followed by fine-grain computational methods to capture the detailed atomic-level structural changes and calculate the thermodynamic properties. Toward these major goals our individual research projects include:
- Development of internal coordinate molecular dynamics simulation methods – GNEIMO
- Development of coarse grained conformational sampling method for G-protein coupled receptors (GPCRs) – GPCRSimKit
- Development of a computational method for designing thermostable mutants for GPCRs – LITiConDesign
- Computational method to map the allosteric communication pipelines in proteins – AlloSteer
- Development of computational methods to identify allosteric sites for drug design in protein-protein complexes
- Application of these methods to design drugs with functional specificity for GPCRs targeting pancreatic cancer and other cancers
- Application of AlloBindSite for disrupting protein-protein interactions
GNEIMO – A Hierarchical Framework for Constrained Molecular Dynamics

- Study of the large-scale conformational dynamics of proteins wherein we showed that the NMR-based conformational ensemble of calmodulin was sampled by GNEIMO method
- Structural refinement of the homology models of proteins
- Ab-initio folding of simple proteins
We are now initiating a collaboration with the Lawrence Berkeley National Laboratory to use the GNEIMO method with the program “PHENIX” to fit models to X-ray crystallography and low-resolution electron microscopy measurements.
The GneimoSim software can be downloaded free of cost for academic use here: http://dartslab.jpl.nasa.gov/GNEIMO/index.php.
Publications related to this project:
- Jain A, Vaidehi N, Rodriguez GA. 1993, Fast Recursive Algorithm For Molecular-Dynamics Simulation, J. Comput. Phys., 106: (2) 258-268.
- Vaidehi N, Jain A, Goddard III, WA. 1996, Constant temperature constrained molecular dynamics: The Newton-Euler inverse mass operator method, J. Phys. Chem., 100: (25) 10508-10517.
- Bertsch RA, Vaidehi N, Chan SI, et al. 1998, Kinetic steps for alpha-helix formation Proteins: Structure, Function and Genetics, 33: (3) 343-357.
- Vaidehi N, Goddard WA. 2000, Domain motions in phosphoglycerate kinase using hierarchical NEIMO molecular dynamics simulations, J Phys. Chem. A 104: (11) 2375-2383.
- Balaraman GS, Park IH, Jain A, Vaidehi N. 2011, Folding of Small Proteins Using Constrained Molecular Dynamics. J. Phys.Chem. B. 115(23):7588-96.
- Park, IH, Wagner J., Jain A, Vaidehi N. 2012, Structure Refinement of Protein Low Resolution Models Using the GNEIMO Constrained Dynamics Method Folding of Small Proteins Using Constrained Molecular Dynamics, J. Phys. Chem. B, 116, 2365-75.
- Jain A, Park IH, Vaidehi N. 2012, Equipartition principle for internal coordinate molecular dynamics, J. Chem. Theory. Comput., 14;8(8):2581-2587.
- Wagner JR, Balaraman GS, Niesen MJ, Larsen AB, Jain A, Vaidehi N. 2013, Advanced techniques for constrained internal coordinate molecular dynamics, J Comput. Chem. ,34(11):904-14
- Gangupomu VK, Wagner JR, Park IH, Jain A, Vaidehi N. 2013, Mapping conformational dynamics of proteins using torsional dynamics simulations, Biophys. J., 104, 1999-2008.
- Jain A, Kandel, S, Wagner, JE, Larsen AB, Vaidehi N. 2013, Fixman compensating potential for general branched molecules, J. Chem. Phys. 139, 244103.
- Larsen AB, Wagner, JR, Jain A, Vaidehi N. 2014, Protein Structure Refinement of CASP target Proteins suing GNEIMO torsional dynamics method, J. Chem. Inf. Model., 24;54(2):508-17.
- Larsen AB, Wagner JE, Kandel S, Salomon-Ferrer R, Vaidehi N, Jain A. 2014, GneimoSim: A modular Internal Coordinates Molecular Dynamics Simulation Package, J. Comp. Chem., 35(31), 2245-2255.
- Vaidehi N, Jain A. 2015. Internal coordinate molecular dynamics: A foundation for multiscale dynamics. J. Phys. Chem. B, 119(4), 1233-1242. (Journal Cover)
GPCRSimKit – A Multiscale Computational Framework for Studying G-Protein Coupled Receptors
G-protein coupled receptors (GPCRs) play an important role in the physiology and pathophysiology of many serious diseases. They form the largest superfamily of drug targets. Since GPCRs are membrane-bound and are highly dynamic, obtaining three-dimensional structural information for GPCRs is a feat requiring a confluence of various biophysical techniques, including computational methods. The crystal structure is a snapshot in the conformational ensemble that the receptor samples in the absence of any stimulant.
We are developing multiscale simulation method suite, GPCRSimKit, that integrates coarse-grain simulation methods with fine-grain techniques. The GPCRSimKit will enable simulation of the dynamics of GPCR conformational ensembles starting from the inactive crystal structures, or refine homology models for drug design. The GPCRSimkit will allow for calculation of the modulation of the potential energy landscape by full, partial and inverse agonists. This platform of computational techniques, will lay a theoretical basis and play a crucial role as more crystal structures of GPCRs are published.
Publications related to this project:
- Vaidehi N., et al 2002, Structure and Function prediction for G-Protein Coupled Receptors, Proc. Natl. Acad. Sci., USA, 99, 12622-12627.
- Bhattacharya S, Hall SE, Li H, Vaidehi N. 2008, Ligand-stabilized conformational states of human β(2) adrenergic receptor: insight into G-protein-coupled receptor activation. Biophys J., 94(6):2027-42.
- Bhattacharya S, Hall SE, Vaidehi N. 2008, Agonist induced conformational changes in bovine rhodopsin: Insight into activation of G-protein coupled receptors, J. Mol. Biol., 382, 539-555.
- Hall SE, Roberts K, Vaidehi N. 2009, Position of helical kinks in membrane protein crystal structures and the accuracy of computational prediction, J. Mol. Graph. & Mod., 27, 944-950.
- Hall SE, Mao A Nicolaidou V, Finelli M, Wise EL, Nedjai B, Kanjanapangka J, Harirchian P. Chen D, Selchau V, Ribeiro S, Schyler S, Pease JE, Horuk R, Vaidehi N. 2009, Elucidation of binding sites of dual antagonists in the human chemokine receptors CCR2 and CCR5. Mol. Pharmacol. 75, 1325-1336.
- Vaidehi N., Pease J, Horuk R. 2009, Modeling Small Molecule Compound Binding to G-Protein Coupled Receptors, Methods Enzymol., 460, 263-288.
- Bhattacharya S, Vaidehi N. 2010, Computational Mapping of the Conformational Transitions in Agonist Selective Pathways of a G-Protein Coupled Receptor, J. Am. Chem. Soc., 132(14):5205-14.
- Bhattacharya S., et al 2010, Allosteric Antagonist Binding Sites in Class B GPCRs: Corticotropin Receptor 1, J Comput Aided Mol Des. 8, 659-74.
- Vaidehi N. 2010, Dynamics and Flexibility of G-protein coupled receptor conformations and its relevance to drug design, Drug Discovery Today, 15, 951-957 – invited review.
- Vaidehi N, Kenakin T. 2010, Conformational Ensembles of Seven Transmembrane Receptors and their Relevance to Functional Selectivity, Curr. Opin. Pharmacol., 10, 775-781- invited review.
- Lam AR, Bhattacharya S, Patel K, Hall SE, Mao A, Vaidehi N. 2011 Importance of receptor flexibility in binding of cyclam compounds to the chemokine receptor CXCR4. J Chem. Inf. Model. 24;51(1):139-47.
- Bhattacharya S, Lam AR, Li H, Balaraman G, Niesen MJ, Vaidehi N. 2013, Critical analysis of the successes and failures of homology models of G protein-coupled receptors, Proteins. 81(5):729-39.
- Bhattacharya S, Vaidehi N. 2012, LITiCon: a discrete conformational sampling computational method for mapping various functionally selective conformational states of transmembrane helical proteins. Methods. Mol. Biol., 914, 167-78.
- Lee S, Bhattacharya S, Grisshammer R, Tate C, Vaidehi N. 2014, Dynamic Behavior of the Active and Inactive States of the Adenosine A2A Receptor. J. Phys. Chem. B. 118(12):3355-65.
- Vaidehi N, Bhattacharya S, Larsen AB. 2014, Structure and dynamics of G-protein coupled receptors, Adv. Exp. Med. Biol., 796, 37-54.
- Muppidi J., Green J. ……Vaidehi N, Staudt L, Cyster J. 2014, Loss of signaling via Gα13 in germinal center Bcell derived Lymphoma, Nature, 516(7530), 254.
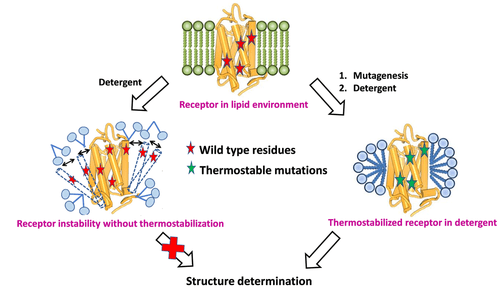
LITiConDesign – A Computational Method for Designing Thermostable Mutants for GPCRs
G-protein coupled receptors are membrane proteins and play an important part in cellular signal transduction. Solving the three-dimensional structures of these proteins is critical – and is lately becoming viable. However, the biggest bottleneck in obtaining sufficient quantities of the pure protein is that GPCRs are conformationally flexible, and hence aggregate at higher concentrations during purification.
A solution to this challenge is to derive thermostable mutants of GPCRs that are amenable to purification techniques. However, the experiments involved in identifying the residue positions that lead to thermostability as well as the thermostable mutants is both expensive and time consuming. There are about 300 mutations that need to be done just to be able to identify positions that lead to thermostability. Our goal is to develop a fast computational screening method, LITiConDesign, to design thermally stable mutants of several GPCRs. We will target class A GPCRs in their agonist- and antagonist-bound structures. This project is in collaboration with Dr. Chris Tate (MRC, Cambridge, UK) and Dr. Reinhard Grisshammer (NINDS).
Publications related to this project:
- Balaraman G., Bhattacharya S., Vaidehi N. 2010, Structural insights into conformational stability of wild type and mutant β1-adrenergic receptor, BioPhys. J., 99(2):568-77.
- Niesen MJM, Bhattacharya S, Grisshammer R, Tate CG, Vaidehi N. 2013, Thermostabilization of the β1-adrenergic receptor correlates with increased entropy of the inactive state, J. Phys. Chem. B., 117, 7283-91.
- Lee S, Bhattacharya S, Grisshammer R, Tate C, Vaidehi N. 2014, Dynamic Behavior of the Active and Inactive States of the Adenosine A2A Receptor. J. Phys. Chem. B. 118(12):3355-65.
- Bhattacharya S, Lee SB, Grisshammer R, Tate CG, Vaidehi N. 2014, Rapid Computational Prediction of Thermostabilizing Mutations for G Protein-Coupled Receptors, J. Chem Theor. & Comp., 10(11), 5149-5160.
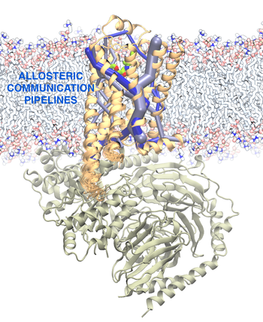
AlloSteer – Development of Computational Method to Identify Allosteric Sites in Proteins for Drug Design
Publications related to this project:
- Bhattacharya S, and Vaidehi N. 2014, Differences in allosteric communication pipelines in the inactive and active states of a GPCR, Biophys. J., 107, 422-34.
- Li H, Kasam V, Tautermann CS, Seeliger D, Vaidehi N. 2014, Computational method to identify druggable binding sites that target protein-protein interactions, J. Chem. Inf. Model. 54(5):1391-400
- Nivedha AK, Tautermann CS, Bhattacharya S, Lee S, Casarosa P, Kollak I. ... Vaidehi N. 2018. Identifying Functional Hotspot Residues for Biased Ligand Design in G-protein-coupled Receptors. Mol. Pharmacol., 93(4), 288-296.