Leo Wang Lab Research
Our laboratory takes a truly translational approach to improving cellular therapies for pediatric cancers. We use protein-focused techniques to understand how cancer-targeting engineered immune cells make cell state and fate decisions, and use that information to design improved adoptive cellular therapies.
This work incorporates synthetic biology, mechanistic and proteomic interrogations, and in vitro and in vivo cellular immunology modeling. In parallel, we incorporate early-phase clinical trials testing these cellular therapies in pediatric patients. These collaborative trials generate unique and valuable correlative data, which in turn informs our bench science. As described below, we have multiple ongoing projects in the lab that focus on chimeric antigen receptor therapies, with the overall goal of improving outcomes for children with cancer.
These projects fall into three main categories:
Learning From Clinical Trial Data to Build Better CAR T Cells
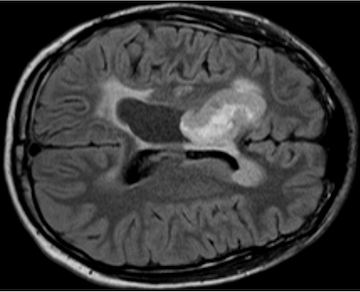
We currently have an open clinical trial, NCT04510051, that tests the intraventricular delivery of IL13Rα2-targeted CAR T cells in children with refractory or recurrent brain tumors. Our trial is one of only a handful that treats children with brain tumors using CAR T cells, and we have seen evidence suggesting that CAR T cell treatment reawakens an endogenous T cell immune response during times of clinical or radiographic improvement.
The overall goal of these projects is to elucidate the mechanisms that drive CAR T-mediated reawakening of the endogenous immune system in pediatric brain tumor patients, and to design and test improved CAR T cells that amplify those drivers to improve endogenous immune reawakening. We use paired single cell RNA sequencing (scRNAseq) and T cell receptor sequencing (TCRseq) of densely-sampled patient samples, anchored to important clinical response categories (e.g., response, stable disease, progression) and combined with multiplex proteomic analysis to extract pathways that drive endogenous antitumor immune reawakening. We then biologically validate candidate pathways in human and murine CAR T cells, evaluating the ability of modified CAR T cells to promote endogenous T cell activation directly, indirectly, or both in in vitro and in vivo model systems.
Cellular and Molecular Characterization of Therapeutic Response and Resistance
We have performed scRNAseq and TCRseq on peripheral blood and cerebrospinal fluid samples from patients on our trial. In collaboration with the Banovich lab, these data-dense samples are analyzed to derive an immune evolutionary landscape tied to discrete response timepoints. This is overlaid with flow cytometry and cytokine analyses to identify biomarkers that correlate with clinical and radiographic improvement or worsening.Endogenous T Cell Clonal Dynamics in the Context of CAR T Therapy
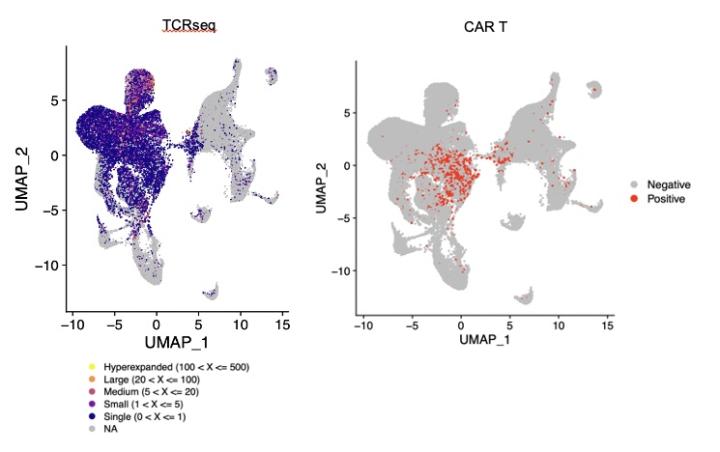
In the first patients treated on our clinical trial, we observed expanded populations of CAR-negative CD8+ T cell clones during treatment. These expanded T cells are not found in peripheral blood or in the CAR T product, indicating that expansion is a consequence of intraventricular CAR T therapy. This finding suggests that CAR T cells are promoting endogenous T cell clonal expansion, which is intriguing because preclinical data demonstrates that CAR T cells can awaken endogenous T cell responses to effect superior control of antigen-negative disease. We are attempting to identify the antigen specificities of expanded endogenous TCRs, as well as to identify molecular drivers of this expansion within endogenous T cells and to identify cellular cues provided by CAR T cells to promote this response. Furthermore, we are modeling CAR T cell and endogenous T cell interactions using murine and human model systems in which both the TCR repertoire and the CAR specificity are specified.
Understanding CAR Signaling and Biology To Improve CAR T Cell Function
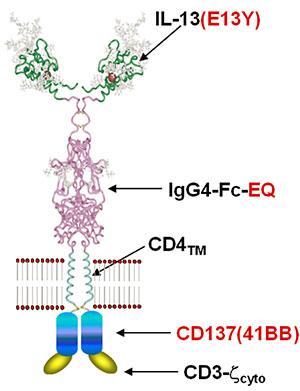
CAR T cells have revolutionized the way that we think about and treat hematologic malignancies, but their application to solid tumors has been much more challenging. Key to improving CAR T cell function for solid tumors is understanding the molecular signaling consequences of antigen recognition. To do this, we study the downstream molecular consequences of receptor ligation using proteomic techniques (flow cytometry, mass spectrometry, Western blot), genomic techniques (RNASeq, RT-PCR), and epigenetic approaches. These studies have identified a number of pathways that may be important for effector CAR T cell activation, persistence, and function, and which differ in their activation dynamics and kinetics across CAR constructs. We are currently investigating the importance of some of these pathways in CAR function.
Mass Spectrometry-Identified Phosphoproteins
We performed bottom-up proteomics on CAR T cells after antigen stimulation, comparing CAR T cells that bear a CD28 costimulatory domain with those bearing a 4-1BB costimulatory domain. This analysis identified differences in NF𝜅B pathway phosphorylation, which we validated using orthogonal approaches to demonstrate that 4-1BB-containing CAR constructs activate the noncanonical NF𝜅B pathway whereas CD28-containing constructs do not (accepted for publication in Cancer Research Communications). Further experiments are ongoing to evaluate the effect of small molecule NF𝜅B inhibitors on CAR T function.
Other candidate phosphoproteins, such as STK10, are also being studied. Additionally, our mass spectrometry analysis identified differential phosphorylation of the CAR itself, although alterations in CAR tyrosine phosphorylation are better assessed using conventional Western blotting. Targeted proteomic analysis of CAR-initiated signaling cascades demonstrated significant differences in the dynamics and kinetics of proximal signaling protein phosphorylation between 4-1BB and CD28 CARs. On the basis of this preliminary data, and of other published studies, we have made specific mutations in the cytoplasmic tail of 4-1BB-containing CARs to test their effects on CAR T cell function.
Epigenetic Modifiers for Preventing CAR T Exhaustion
Understanding that T cell exhaustion is driven by both transcriptional and epigenetic programs, we sought to reverse or prevent CAR T exhaustion using epigenetic reprogrammers. Our preliminary data demonstrate that IL13Rα2-targeted CAR T manufactured in the presence of specific modifiers provides superior tumor control than control cells, in part due to increased proliferative capacity, decreased differentiation, and decreased exhaustion. Ongoing work evaluates the epigenetic and transcriptional effects of this treatment and will identify other epigenetic modifiers that can be used in CAR T manufacturing.
Synthetic Biology Approaches to Improving Chimeric Antigen Receptor (CAR) T Cell Function
CAR T cell development has in large part been driven by landmark advances in synthetic biology, and this approach is likely to improve the function of solid tumor targeted CAR T cells. Solid tumors remain relatively resistant to CAR T cell therapy, for a variety of reasons; these include antigen heterogeneity, antigen escape, and a uniquely immunosuppressive tumor microenvironment. Introducing additional genetic circuits to address these important obstacles to CAR T cell function is an area of great interest. In our lab, we have undertaken several projects to provide additional signaling that will augment or focus CAR T cell function.
Membrane Bound Cytokines
To overcome exhaustion, many groups are providing adjunctive cytokine stimulation. We have collaborated with the Priceman lab to study membrane bound IL12 constructs, which we have shown improve CAR T cell persistence, homing, and efficacy. Additionally, we are developing membrane bound cytokine partial agonist constructs to expand less-differentiated, more active CAR T cells in both the antitumor T effector setting and in the immunomodulatory T regulatory setting (with the Montero lab).
CAR T Brakes
Many solid tumor-associated antigens are heterogeneously expressed, and targeting them leads to selective pressure, with resulting outgrowth of antigen-negative disease. However, most antigens with universal expression on tumor cells are also expressed on normal tissue. To overcome antigen heterogeneity, many labs are adopting bi- or tri-specific targeting approaches; others are using adaptive or SynNotch approaches to trigger CAR switching at particular times or in particular places. Our approach adds inhibitory CARs that, upon recognition of a “safe harbor” antigen, activate phosphatases to dampen target antigen-mediated CAR activation and prevent CAR T cell killing. In this way, we can broaden the repertoire of target antigens addressable with CAR T cells, while also providing an additional layer of safety to prevent killing of normal cells.